


Qué es?
La Deficiencia de piruvato quinasa es un error congénito del metabolismo de los hematíes (eritrocitos o glóbulos rojos), que causa una anemia hemolítica crónica.
Este trastorno provoca que los glóbulos rojos se destruyan más rápido de lo que la médula ósea puede producirlos.
Esto implica una anemia crónica en el niño que se va acentuando día a día, hasta que la situación requiere de una transfusión de sangre.
¿Qué tratamiento tiene?
En casos graves como Bernat, la vida del paciente desde el periodo neonatal está comprometida, y requiere transfusiones frecuentes. Esta es la única solución hasta llegar a una edad más avanzada (5 años), donde se podrá realizar una esplenectomía. En caso de que así no mejore, la única solución podría ser un trasplante de médula ósea.
En los casos leves, muchas veces no requiere tratamiento.
El déficit de Piruvato Quinasa, al detalle

El Piruvato Quinasa es una enzima que proporciona energía a los glóbulos rojos (o hematíes). Estos tienen una función muy importante, que es la de transportar el oxígeno a todos los tejidos del cuerpo.
Pero cuando hay una falta de Piruvato Quinasa, los glóbulos rojos se destruyen. Y por otra parte, cuando se forman nuevos hematíes, todos están afectados con esta deficiencia enzimática.

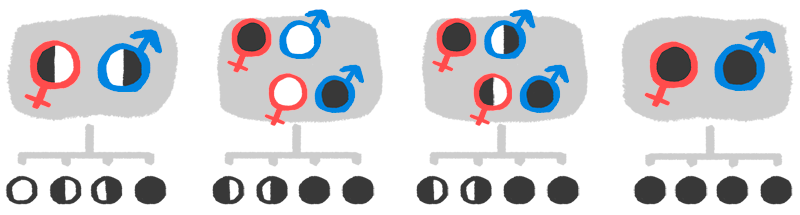
El Déficit de Piruvato Quinasa es una enfermedad denominada como rara y considerada como minoritaria. Es una anemia hemolítica crónica, que el enfermo ha heredado genéticamente por parte del padre y de la madre. Así, se pueden dar hasta cuatro casos para transmitir la enfermedad:
- Los dos padres sean portadores pero no afectados.
- Uno sea enfermo y el otro completamente sano.
- Uno de los dos sea portador no afectado, y el otro portador afectado.
- Ambos sean enfermos, y por lo tanto, portadores.
Esta anemia también provoca una destrucción masiva de los glóbulos rojos, porque tienen un bajo número de hemoglobina (proteína responsable del transporte del oxígeno). El tratamiento para los casos más severos, como es el de Bernat, es hacer constantes transfusiones de sangre para seguir en vida.
En estos casos tan graves de la enfermedad los hematólogos, dependiendo de cada caso, aconsejan la extirpación del bazo a pesar de los riesgos que conlleva. Y si esto funciona, puede existir la posibilidad de alargar un poco los intervalos entre transfusiones. En el caso de Bernat, la idea es llevarlo a cabo hacia los cinco años de edad.
Por otra parte, también se puede llegar a plantear el trasplante de la médula ósea como forma de cura definitiva. Esta opción, se enfoca en casos que la extirpación del bazo no ha servido y el enfermo tiene una necesidad bastante constante de transfusiones de sangre. Por otra parte, también depende de cada caso y la capacidad médica del enfermo respecto a los riesgos que hay a lo largo del proceso e intervención.
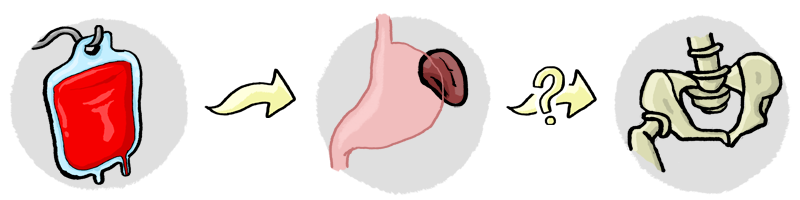
Actualmente, debido a la gravedad que sufre Bernat, estamos preparándonos para esta opción. No es seguro, pero hay muchas posibilidades. Por este motivo, estamos a esperas que nos aprueben una fecundación in vitro en la cual se elijan los embriones sanos y compatibles de médula ósea con Bernat. Pues debido a sus características médicas, si recibiese una médula de un donante compatible que no fuera un hermano, se elevaría de forma exagerada la tasa de mortalidad.
Por último, a fecha de hoy, se está trabajando en lo que llaman Terapia Génica como alternativa al trasplante de la médula ósea. Es una gran esperanza, pero está en fase experimental.
Fuente informativa: Guía Metabólica Sant Joan de Déu
Más información en la Red
- La Guía Metabólica de Sant Joan de Déu
- Librería Nacional de Medicina
- Organización Mexicana de Enfermedades Raras
Links relacionados
